|
|
|
|
|
|
|
|
| Leucodistrofias.
São doenças progressivas da mielina
ou de suas células formadoras (oligodendrócitos), devidas
a erros inatos do metabolismo de causa genética e, freqüentemente,
envolvendo os lisossomos ou os peroxissomos. Geralmente apresentam-se já
na infância como doenças dismielinizantes, no sentido de que
a mielina formada é defeituosa desde a origem, do ponto de vista
estrutural ou metabólico, ou desmielinizantes, em que a mielina
é destruída por acúmulo de produtos metabólicos
como os sulfátides (na leucodistrofia metacromática)
ou os glicolípides (na leucodistrofia de
células globóides).
O diagnóstico é estabelecido pela apresentação clínica, exames de neuroimagem, exames bioquímicos do sangue, urina ou líquor, eletroneurografia e análises genéticas. É raro que leucodistrofias sejam encontradas em material cirúrgico (pois não são normalmente biopsiadas), exceto em casos não diagnosticáveis pelos métodos acima, como na leucodistrofia de Alexander. Clique para detalhes sobre adrenoleucodistrofia : textos (1) (2), neuroimagem, neuropatologia. leucodistrofia metacromática : texto, neuroimagem, neuropatologia. leucodistrofia de células globóides ou doença de Krabbe : texto, neuropatologia. leucodistrofia de Pelizaeus-Merzbacher : texto, neuropatologia. leucodistrofias sudanófilas ou ortocromáticas : texto, neuropatologia. leucodistrofia de Alexander : texto, neuropatologia. leucodistrofia de Canavan : texto, neuroimagem, neuropatologia. leucodistrofia de van der Knaap : texto, neuroimagem. leucodistrofia associada a distrofia muscular congênita por deficiência de merosina : texto, neuroimagem, biópsia muscular. As figuras
deste texto têm links diretos às páginas originais.
Clique nas figuras para ir às páginas.
|
| Adrenoleucodistrofia.
É a leucodistrofia mais comum, ligada ao sexo e com 3 manifestações clínicas distintas.
Clinicamente, pacientes com as formas cerebrais mostram déficits cognitivos, auditivos e visuais progressivos, com óbito em alguns anos. A adrenomieloneuropatia se caracteriza por paraparesia e distúrbios esfincterianos progressivos. Exames de imagem. Há desmielinização progressiva da substância branca dos hemisférios cerebrais, geralmente iniciando-se nos lobos occipitais e progredindo rostralmente aos lobos frontais. Há severo envolvimento das estruturas da linha média, como corpo caloso, fórnix, nervos, quiasma e tratos ópticos. Áreas desmielinizadas mostram hipossinal em T1 e hipersinal nas seqüências com TR (tempo de repetição) longo, T2 e FLAIR. É comum impregnação por contraste na margem das regiões desmielinizadas, correspondendo na neuropatologia ao acúmulo de macrófagos xantomatosos e infiltrado inflamatório linfocitário perivascular. |
| Adrenoleucodistrofia. Ressonância magnética. Masc. 6 a. Extensa desmielinização da substância branca occipital e parietal, do trato piramidal no mesencéfalo e ponte (setas vermelhas) e relativa preservação das regiões frontais e joelho do corpo caloso. Com contraste, impregnação forte nas margens das áreas desmielinizadas. | ||
| AXIAIS, T2 | T1 COM CONTRASTE | |
 |
 |
 |
 |
||
| CORONAL, T2 | T1 COM CONTRASTE | SAGITAL, T1 SEM CONTRASTE |
 |
 |
 |
| Masc. 11 a. Lesões da mielina predominam nas regiões posteriores dos hemisférios cerebrais, com atrofia da substância branca, hipossinal em T1 e hipersinal em T2 e FLAIR. Dilatação ventricular ex-vacuo, maior nos cornos posteriores dos ventrículos laterais. Nos cortes sagitais na linha média, alteração de sinal no terço posterior do corpo caloso, contrastando com preservação dos 2/3 anteriores. | ||
| T1 SEM CONTRASTE | FLAIR | T2 |
 |
 |
 |
| CORONAIS, FLAIR | SAGITAIS, T1 SEM CONTRASTE | ||
 |
 |
 |
 |
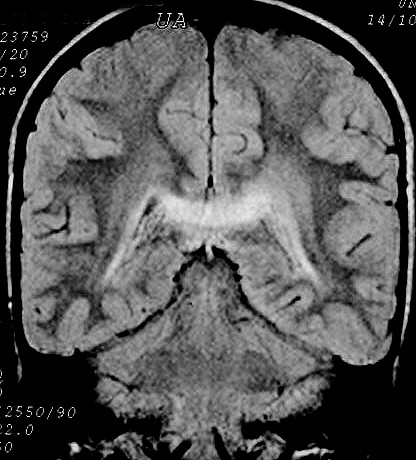
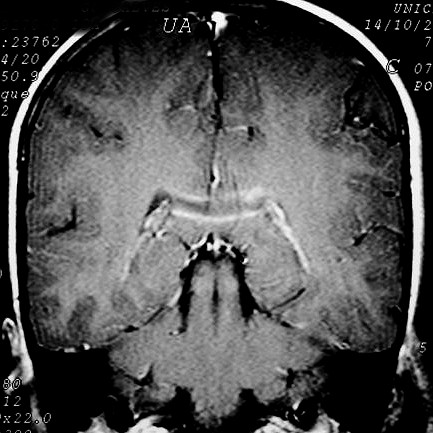
| Masc. 10 a. Irmão do paciente acima. Caso frusto. Desmielinização do esplênio do corpo caloso, fórceps maiores e substância branca parieto-occipital, tênue impregnação por contraste na periferia da região posterior do corpo caloso. | |||
| FLAIR | T2 | FLAIR | T1 COM CONTRASTE |
 |
 |
 |
 |
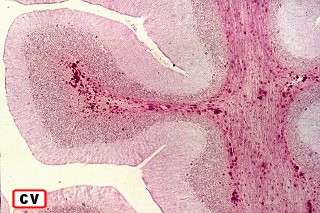
| Adrenoleucodistrofia.
Neuropatologia.
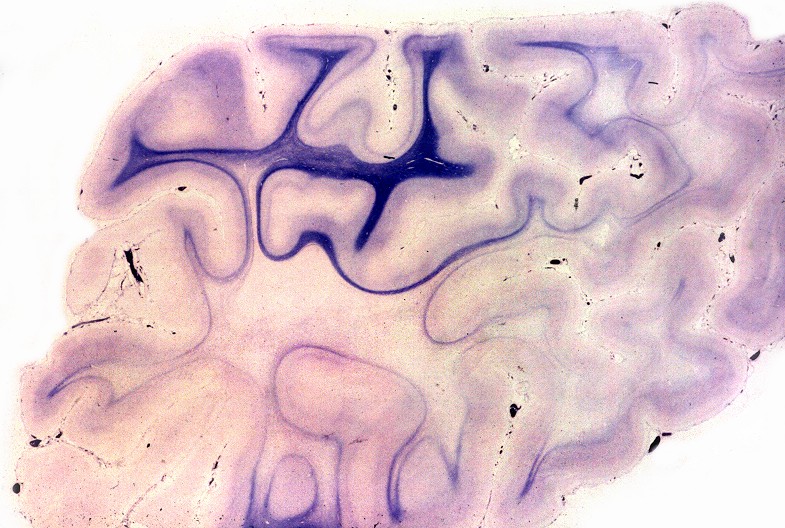
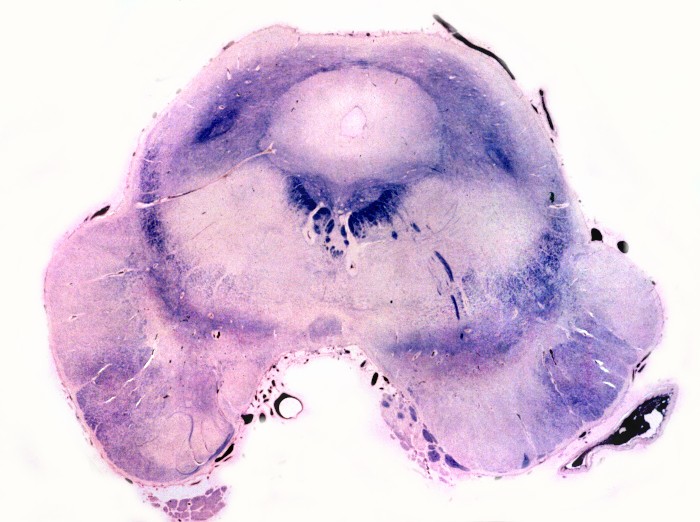
Há lesão difusa da substância branca dos hemisférios cerebrais com desmielinização confluente e simétrica. Costuma haver predomínio nas regiões posteriores nas fases iniciais, avançando aos lobos frontais com a progressão da doença. As fibras em U subcorticais são poupadas. No cérebro predominam macrófagos xantomatosos que fagocitaram restos de mielina degenerada. Chamam a atenção também infiltrados linfocitários perivasculares compostos predominantemente por células T. Há gliose reacional nas áreas desmielinizadas que, no longo prazo, pode chegar à cavitação. Na mielopatia, há degeneração dos tratos longos, especialmente das colunas posteriores e dos tratos corticospinais, mais notadamente na medula cervical. Não há inflamação ou é limitada a células esparsas. Acometimento de nervos periféricos é variável, discreto e sem processo inflamatório. Corresponde histologicamente a uma neuropatia desmielinizante, com baínhas de mielina anormalmente delgadas e desmielinização segmentar em preparações com teasing. Outros órgãos. É característico o envolvimento das camadas fasciculada e reticular do córtex adrenal, células de Leydig dos testículos e nas células de Schwann dos nervos periféricos. As células do córtex adrenal ficam volumosas, com contorno arredondado, citoplasma róseo abundante e finamente granuloso. Em microscopia eletrônica, inclusões intracitoplasmáticas, descritas como lamelares e lineares, lembram fendas limitadas por membranas ou lamelas finas (cerca de 3 nm de espessura) separadas por espaço claro de 4-10 nm. Pode haver insuficiência crônica da supra-renal, ou doença de Addison, incluindo hiperpigmentação cutânea. |
| Cérebro, coloração de Loyez para mielina. Lobo frontal. Perda completa da mielina da substância branca, exceto das fibras subcorticais em U. | Lobo occipital. | Cerebelo, desmielinização central em torno do núcleo denteado |
 |
 |
 |
| Mesencéfalo. Desmielinização do pedúnculo cerebelar superior e dos tratos piramidais. | Ponte. Desmielinização dos tratos piramidais na base da ponte. | Bulbo. Desmielinização dos tratos piramidais (pirâmides bulbares). |
 |
 |
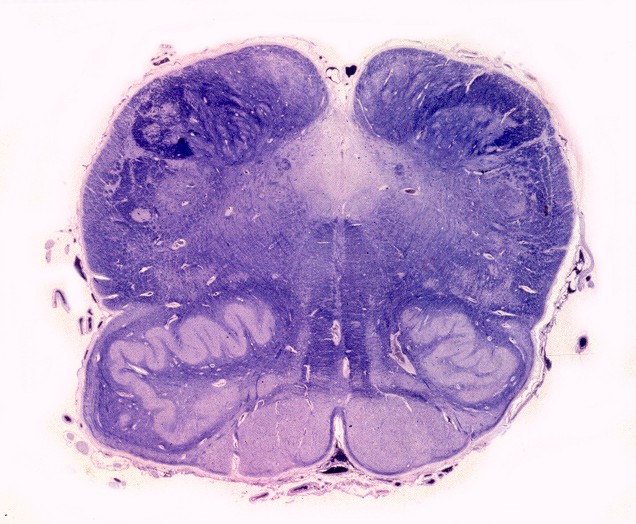 |
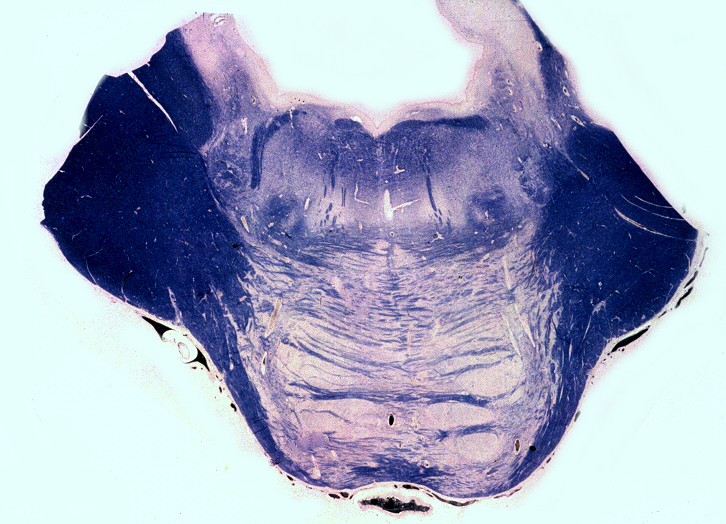
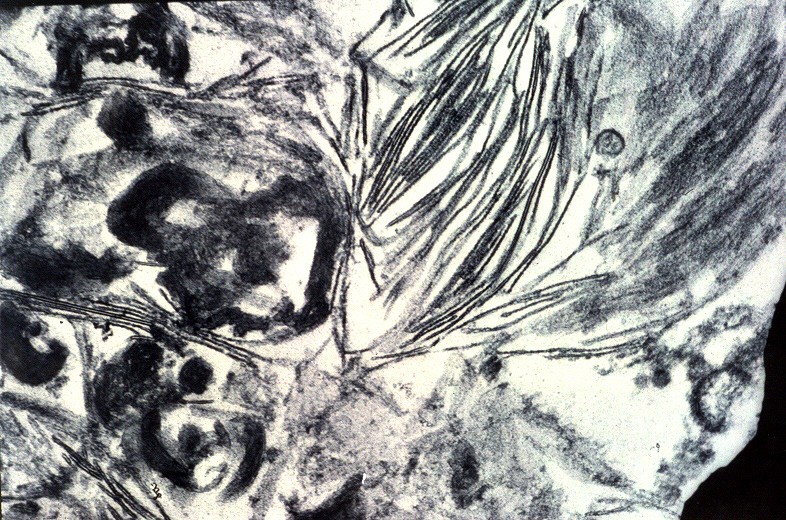
| Medula espinal. Desmielinização dos tratos piramidais no funículo lateral | Supra-renal, camada cortical. Células com citoplasma globoso e material armazenado. Outro caso | Idem, microscopia eletrônica. |
 |
 |
 |
| Adrenoleucodistrofia.
Genética e patogênese.
O gene para adrenoleucodistrofia (ALD) está mapeado ao cromossomo
Xq28. Mais de 400 mutações já foram identificadas,
sem correlação com o fenótipo. O gene codifica um
membro do chamado ATP-binding cassette, subfamily D, member 1'
ou ABCD1, localizado nos peroxissomos e envolvido no transporte dos ácidos
graxos de muito longa cadeia para o interior destas organelas para catabolismo.
Na ausência de ABCD1, esses ácidos graxos se acumulam no sangue,
cérebro e células corticais adrenais, células de Leydig
e células de Schwann.
Normalmente, os ácidos graxos de cadeia muito longa são metabolizados nos peroxissomos a ácidos graxos de cadeia mais curta, que são então exportados ao citosol onde podem ser usados na biossíntese de outros lípides complexos. Entre estes estão os plasmalógenos, essenciais na composição das bainhas de mielina. A falta destes leva à degeneração da mielina, com acúmulo de macrófagos xantomatosos e perda secundária dos axônios na substância branca. Peroxissomos. São organelas citoplasmáticas encontradas em todas células eucarióticas, que contêm enzimas oxidativas como a catalase e a urato-oxidase. Estas estão em concentrações tão altas que podem chegar a formar inclusões cristalóides visíveis em microscopia eletrônica. As enzimas nos peroxissomos usam oxigênio molecular para remover átomos de hidrogênio de certos substratos orgânicos específicos. A reação resultante produz peróxido de hidrogênio (H2O2). Por exemplo, a catalase presente nos peroxissomos usa H2O2 gerado por outras enzimas da organela para oxidar vários substratos, como fenóis, ácido fórmico, formaldeído e álcool. A reação é particularmente importante em hepatócitos e células renais, onde os peroxissomos destoxificam várias moléculas que entram a corrente circulatória. Como exemplo, cerca de 25% do álcool ingerido em bebidas é oxidado a acetaldeído por esta reação. Quando H2O2 se acumula em excesso, a catalase o converte em água com liberação de O2. Outra importante função das reações oxidativas nos peroxissomos é a quebra de moléculas de ácidos graxos, particularmente os de cadeia muito longa (very long chain fatty acids ou VLCFAs). No processo conhecido como beta-oxidação, a cadeia alifática dos ácidos graxos é encurtada em blocos de 2 carbonos por vez para conversão em acetil CoA. Este é exportado dos peroxissomos para o citosol para reutilização em reações biossintéticas. Em mamíferos, a beta-oxidação ocorre tanto nos peroxissomos quanto nas mitocôndrias. Em fungos e vegetais, esta reação essencial só ocorre nos peroxissomos. Plasmalógenos. Uma função biossintética essencial dos peroxissomos de animais é catalizar as primeiras reações na síntese dos plasmalógenos, que são a classe mais abundante de fosfolípides na mielina. Deficiência nos plasmalógenos causa severas anormalidades na formação desta. Os plasmalógenos são fosfogliceróis (compostos da molécula básica do glicerol com um aminoálcool esterificado com ácido fosfórico no carbono 3), e que no carbono 1 contêm um álcool insaturado em ligação éter, em vez de um ácido graxo em ligação éster. Dependendo do aminoálcool no carbono 3, os plasmalógenos pertencem a 3 classes fosfatidalcolinas, fosfatidaletanolaminas e fosfatidalserinas. Diagnóstico
diferencial. O
principal diagnóstico diferencial da adrenoleucodistrofia é
a esclerose múltipla severa de início na infância,
que mostra desmielinização confluente e também pode
ter infiltrados linfocitários perivasculares. É de
interesse histórico que, dos casos de Schilder de leucodistrofia
inflamatória descritos entre 1912 e 1924, um era adrenoleucodistrofia,
outro era esclerose múltipla aguda severa e o terceiro, panencefalite
esclerosante subaguda (causada pelo vírus do sarampo). Isto realça
as feições superponíveis das doenças inflamatórias
da substância branca.
|
| Leucodistrofia
metacromática.
É uma doença de armazenamento autossômica recessiva causada pela deficiência da enzima lisossomal aril-sulfatase, responsável pela degradação dos sulfátides. Isto leva ao acúmulo desta substância em macrófagos no sistema nervoso central e em células de outros órgãos. O gene para aril-sulfatase foi mapeado ao cromossomo 22q13, e mais de 40 mutações já foram identificadas. Descrevem-se formas infantil tardia (início entre 1 e 3 anos de idade), juvenil (início na infância tardia / adolescência) e adulta (início entre 20 e 30 anos). A incidência na população é entre 1:40.000 e 1:100.000. Homozigose para as mutações abole a função da enzima completamente, causando as formas infantis. Mutações missense reduzem a atividade enzimática a 3 5 % do normal, e são responsáveis pelas formas adultas. Pacientes heterozigotos para uma mutação null e uma mutação missense podem desenvolver o fenótipo juvenil. Clinicamente, na forma infantil há ataxia, disartria, disfagia, disfunção cognitiva, com progressão ao óbito após vários anos. Na forma adulta predomina quadro psiquiátrico, com alterações de comportamento e psicose, devido ao predomínio do acometimento frontal. |
| Leucodistrofia metacromática. Ressonância magnética. Masc. 7 a. Hipersinal em T2 e FLAIR, e correspondente hipossinal em T1, afetando difusamente a substância branca profunda e superficial dos hemisférios cerebrais, corpo caloso e cápsula interna. | |||
| AXIAIS, T2 | |||
 |
 |
 |
 |
| T1 SEM CONTRASTE | CORONAIS, T1 COM CONTRASTE | ||
 |
 |
 |
 |
| Fem. 30 a. Diagnóstico presumível de leucodistrofia metacromática, forma do adulto, sem comprovação histológica. Desmielinização avançada da substância branca dos hemisférios cerebrais. | |||
| AXIAIS, T2 | FLAIR | T1 SEM CONTRASTE | |
 |
 |
 |
 |
| CORONAIS, T2 | T1 SEM CONTRASTE | ||
 |
 |
 |
 |
| Para outro caso presumível de leucodistrofia metacromática, forma do adulto, clique. | |||
| Leucodistrofia
metacromática. Neuropatologia.
Nos estágios precoces, as lesões predominam nos lobos frontais.
Nos estágios avançados, há desmielinização
confluente e simétrica com preservação das fibras
em U e sem diferenças significativas em relação aos
outros tipos de leucodistrofia.
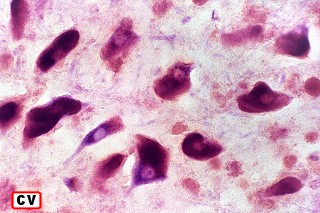
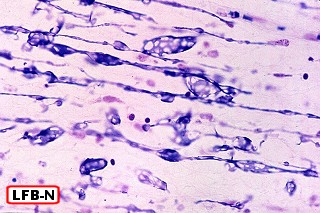
Na microscopia, é característico o acúmulo de material metacromático em cérebro, nervos periféricos, epitélio da vesícula biliar, ductos biliares e túbulos renais. A desmielinização severa do cérebro não se acompanha de reação inflamatória. Os macrófagos contêm abundante material eosinófilo e PAS positivo no citoplasma, que é caracterizado pela reação metacromática, em que a cor usual do corante muda. P. ex. o material corado por azul de toluidina fica róseo, e o cresil violeta ácido torna-se marrom. A mudança de cor se deve ao sulfátide presente na substância armazenada. Para mais sobre esfingolípides, cerebrósides e sulfátides, clique. |
||
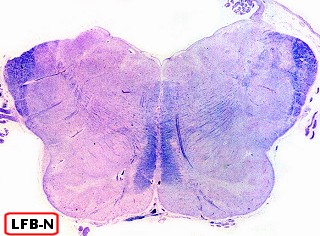
| Cortes de celoidina de lobos cerebrais e cerebelo, corados por LFB-Nissl, mostrando extensa desmielinização da substância branca | Idem, tronco cerebral e medula espinal | Macrófagos contendo substância metacromática (sulfátide) na substância branca das folhas cerebelares e em neurônios do núcleo denteado. Cresil-violeta |
 |
 |
 |
| Acúmulo de substância metacromática (sulfátide) em neurônios e macrófagos no tronco cerebral (foto, neurônios motores do núcleo do facial) | Armazenamento em macrófagos no córtex cerebral. Neurônios não acumulam | Substância branca cerebral : rarefação axonal, alterações degenerativas nas baínhas de mielina, armazenamento em macrófagos |
 |
 |
 |
| Outro caso. | ||
| Corte de celoidina de córtex cerebral e substância branca, corado por azul de toluidina, mostrando extensa desmielinização da substância branca e acúmulo de substância metacromática no limite córtico-subcortical | Macrófagos contendo substância metacromática na substância branca cerebelar, córtex normal. Azul de toluidina | Idem, PAS |
 |
 |
 |
| Acúmulo de substância metacromática (sulfátide) em nervo periférico. Azul de toluidina | Armazenamento de substância metacromática (sulfátide) em fígado. Azul de toluidina | Armazenamento de substância metacromática (sulfátide) em células tubulares renais. Azul de toluidina |
 |
 |
 |
| Leucodistrofia
de células globóides ou de Krabbe.
É uma doença autossômica recessiva que se manifesta no primeiro ano de vida. As crianças nascem normais, mas aos 4 a 6 meses desenvolvem sintomas neurológicos, como falta de aquisições motoras, hiperirritabilidade, hipersensibilidade a estímulos, hipertonia, atrofia óptica e severa deterioração mental. Dificilmente a sobrevida ultrapassa 2 anos. A incidência da forma infantil é de 1:100.000 a 1:200.000. Há envolvimento do sistema nervoso central e de nervos periféricos, mas não de outros órgãos. Reconhecem-se raras formas tardias: juvenil (início aos 3 a 10 anos) e adulta (início na 3ª. a 5ª. décadas). O gene foi mapeado ao cromossomo 14q25-31. Mais de 65 mutações já foram descritas. Há deficiência da enzima lisossomal galactosil-ceramidase (ou galactocerebrosídeo beta galactosidase) levando a acúmulo do intermediário galactosil-esfingosina ou galactocerebrosídeo, ou psicosina, que é tóxico a neurônios, oligodendrócitos e células de Schwann. Radiologicamente há lesões periventriculares simétricas hiperintensas em T2. Em estágios avançados pode haver atrofia cerebral. |
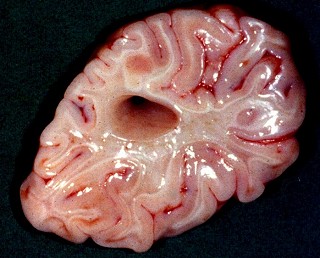
| Neuropatologia.
Há desmielinização simétrica da substância
branca dos hemisférios cerebrais, que adquire coloração
acinzentada e consistência mais firme (devida à gliose). Na
microscopia há perda virtualmente total da mielina profunda, com
relativa preservação das fibras em U subcorticais.
O material lipídico (galactocerebrosídeo) decorrente da falta da enzima é PAS-positivo e acumula-se em macrófagos na substância branca. Estas células são caracteristicamente globóides, por vezes multinucleadas, e tendem a localizar-se em torno de vasos. Há grande declínio do número de oligodendrócitos e hiperplasia e hipertrofia reacionais dos astrócitos (gliose). Reação inflamatória é mínima ou ausente. Em microscopia eletrônica, os macrófagos contêm inclusões membranáceas alongadas, tubuliformes, com contorno irregular. Nervos periféricos
mostram proliferação dos fibroblastos, fibrose, desmielinização
segmentar, formação de bulbos de cebola (por hiperplasia
reacional das células de Schwann), e macrófagos PAS-positivos
perivasculares.
|
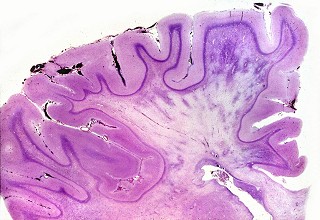
||
| Caso 1, macroscopia a fresco | Caso 2, lobo occipital, Loyez | Ponte, Loyez |
 |
 |
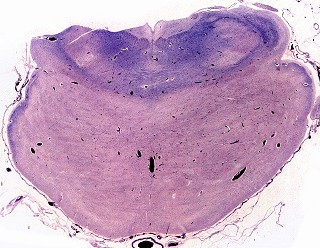 |
| Bulbo, Weigert-Pal | Cerebelo, Loyez | Caso 3, hemisfério cerebral e corpo caloso, PTAH |
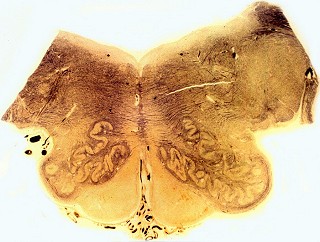 |
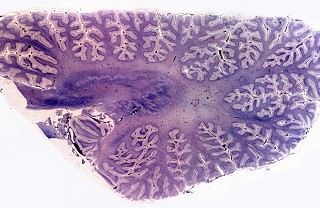 |
 |
| Microscopia. Caso 4 | ||
| HE. Células globóides (macrófagos) na substância branca | LFB - Nissl. Idem, ausência de bainhas de mielina | Glees. Pobreza de axônios na substância branca. |
 |
 |
 |
| Holzer. Gliose - abundantes fibrilas gliais | Cajal. Astrócitos hipertróficos na substância branca | ME. Perfis tubuliformes no citoplasma de macrófagos |
 |
 |
 |
| Leucodistrofia
de Pelizaeus-Merzbacher.
O nome doença de Pelizaeus-Merzbacher (leucodistrofia hipomielinizante) já foi usado para descrever 5 tipos de leucodistrofias sudanófilas, com quadros clínicos e anormalidades anatômicas semelhantes, mas diferindo pela idade de início, taxa de progressão e transmissão genética. A forma clássica tem herança recessiva ligada ao sexo. Pacientes apresentam já nos primeiros meses com movimentos nistagmóides dos olhos, retardo do desenvolvimento motor, espasticidade e movimentos involuntários. O curso é lento e pode parecer estático, levando ao diagnóstico de paralisia cerebral. Morte ocorre no fim da adolescência ou início da idade adulta. Na forma conatal a herança pode ser recessiva ligada ao sexo ou autossômica, assim também meninas podem ser afetadas. Os sinais podem estar presentes já ao nascimento ou iniciar-se precocemente no período pós-natal. Além dos movimentos nistagmóides e hipercinesia extrapiramidal, desenvolve-se espasticidade, atrofia óptica e convulsões, com morte no início da infância. Todas formas
da doença parecem resultar de anormalidades (duplicação
ou deleção) do gene PLP1 no cromossomo Xq22, codificando
a proteína proteolipídica 1 (proteo-lipid protein, PLP),
a principal proteína das membranas da baínha de mielina do
sistema nervoso central, e sua isoforma DM20. Há mais de 40 mutações
identificadas, a maioria levando a duplicação do gene. Outras
são mutações pontuais.
|
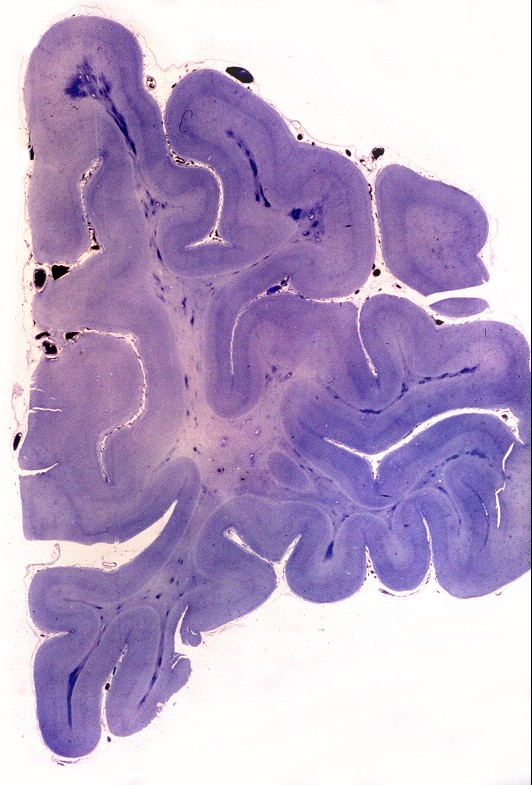
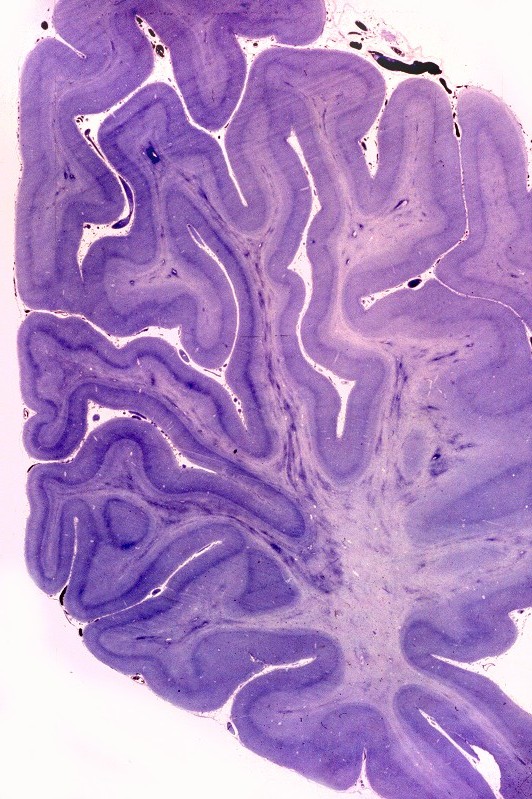
| Neuropatologia. Desmielinização difusa da substância branca, com ilhotas de preservação focal da mielina perivascular, dando um aspecto tigróide característico. Há severa perda dos oligodendrócitos, com apoptose, acompanhada por gliose reacional. Há forte redução do peso cerebral relativo a controles da mesma idade. Nervos periféricos não são afetados, porque as proteínas estruturais da mielina nestes são outras (PMP-22). | ||
| Lobo frontal, Loyez. Caráter 'tigróide', com desmielinização maciça da substância branca e preservação de pequenas ilhotas de mielina perivascular. | Lobo occipital, Loyez. Aspecto semelhante. | Lobo temporal, núcleos da base, Loyez. Aspecto semelhante, desmielinização da cápsula interna. |
 |
 |
 |
 |
||
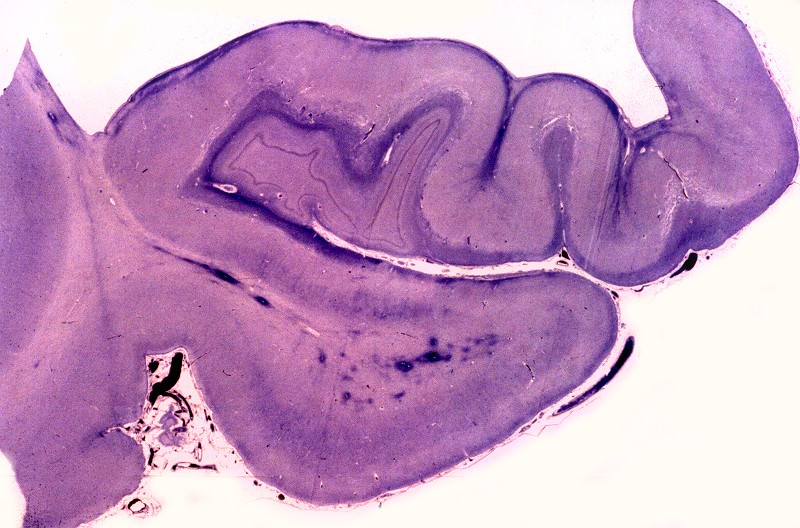
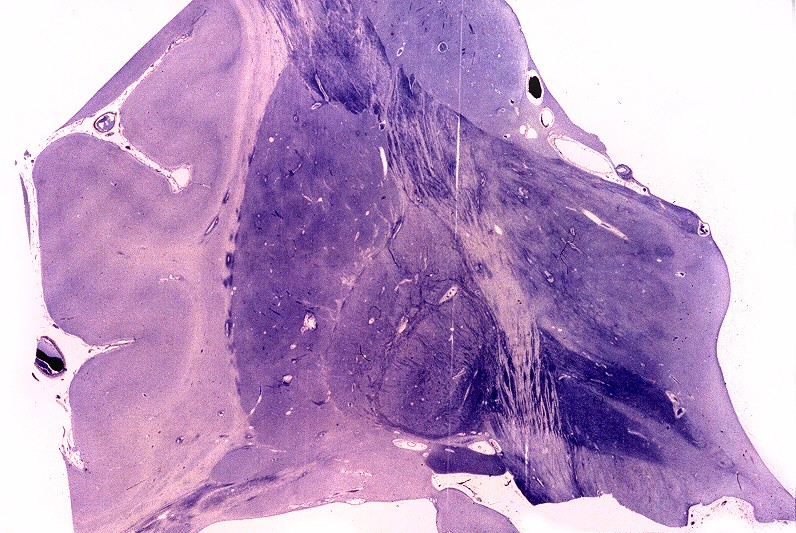
| Núcleos da base, Weigert-Pal. Desmielinização da cápsula interna | Tronco cerebral, Loyez. Desmielinização do trato piramidal | Substância branca cerebral, Loyez. Desmielinização maciça da substância branca e preservação de pequenas ilhotas de mielina perivascular |
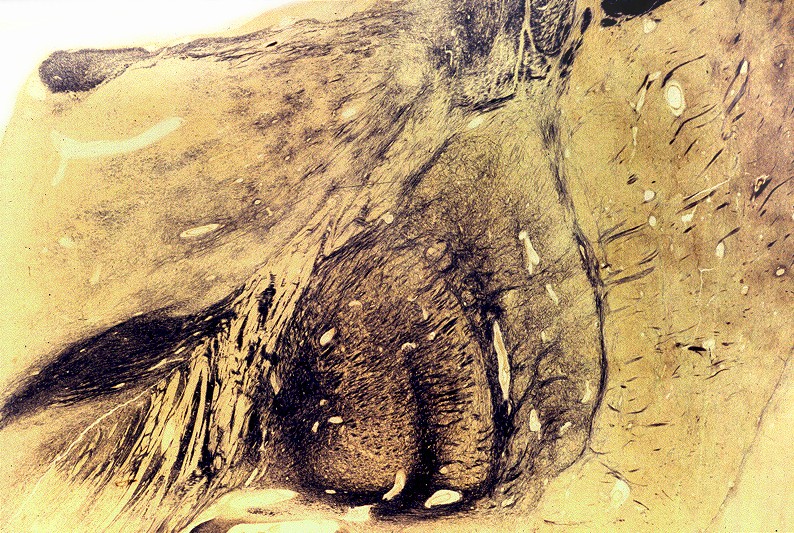 |
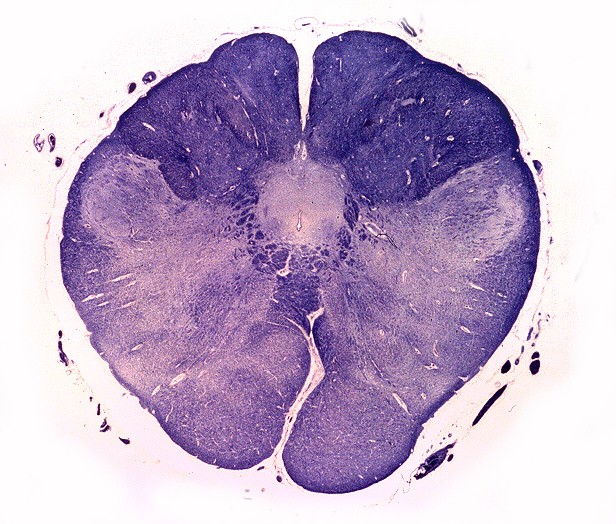 |
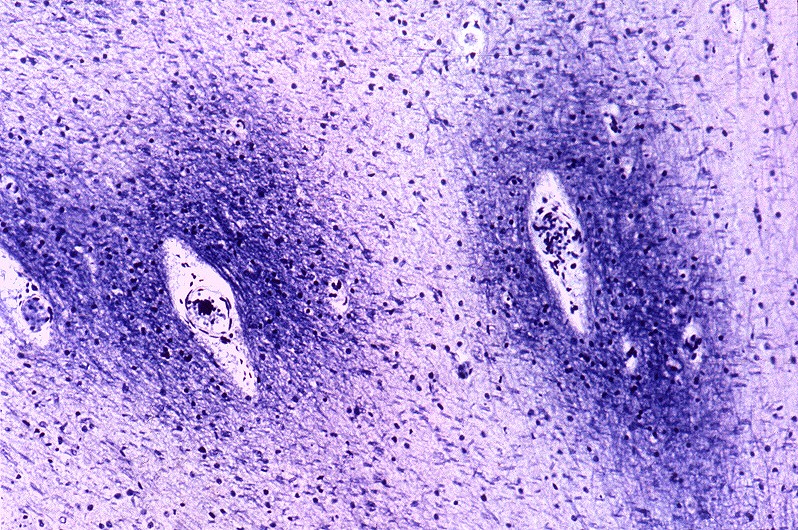 |
| Leucodistrofias
sudanófilas ou ortocromáticas.
Estas constituem um grupo heterogêneo e temporário, em que vários casos esporádicos ou familiais são incluídos por falta de diagnóstico genético preciso. Não correspondem a uma entidade nosológica definida ou separada. O critério é puramente anátomo-patológico, na falta de definição patogenética. Alguns casos são infantis, apresentando-se antes da idade de 5 anos, com curso rápido. Há também casos em adultos com evolução arrastada, e representam as leucodistrofias mais comuns do adulto. As lesões envolvem só o sistema nervoso central, com desmielinização difusa, mal definida e irregular, tendendo a predominar nos lobos frontais e causando sintomas psiquiátricos. A reação macrofágica é discreta, há freqüente envolvimento axonal e não há reação inflamatória. |
| Neuropatologia. Caso 1. | ||
| Macro. Atrofia e cavitação da substância branca frontal e temporal, dilatação ex-vacuo dos ventrículos laterais e III ventrículo | LFB-Nissl. Extensa desmielinização da substância branca frontal | Desmielinização da substância branca das cápsulas externa e extrema e do córtex da ínsula, preservação da cápsula interna e comissura anterior |
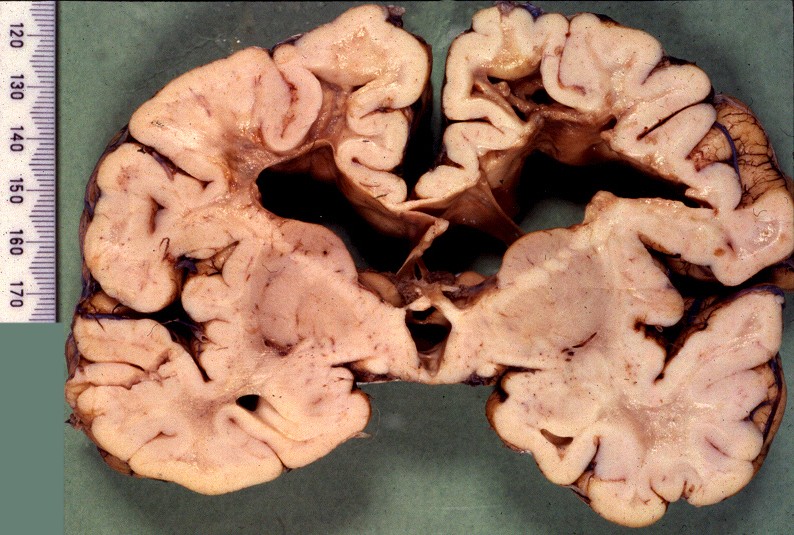 |
 |
 |
| Extensa desmielinização da substância branca temporal e dilatação ex-vacuo do corno inferior do ventrículo lateral | Mesencéfalo - perda parcial da mielina das bases dos pedúnculos cerebrais | Medula espinal - desmielinização parcial dos tratos piramidais no funículo lateral |
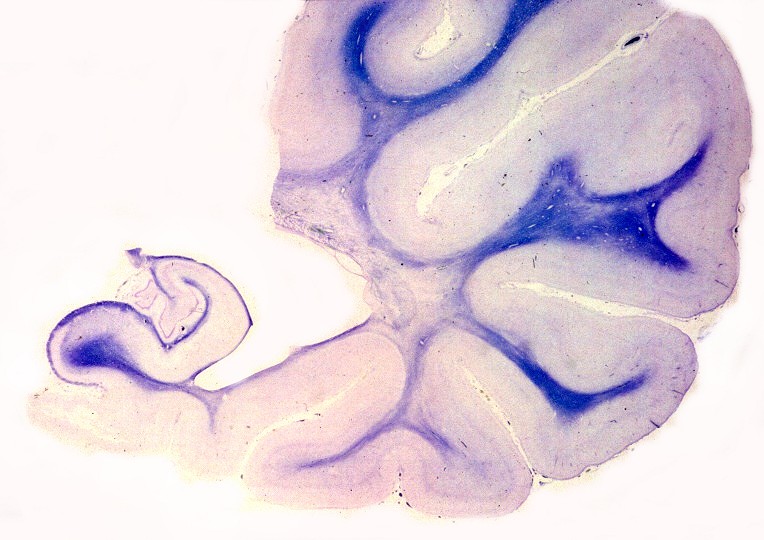 |
 |
 |
| Caso 2. | ||
| Macro. Cor acinzentada da substância branca dos hemisférios cerebrais (aqui, lobos frontais) | Corte frontal, Loyez. Palidez da mielina da substância branca frontal, desmielinização mais intensa no lobo temporal | Lobos temporal e occipital, LFB-Nissl. Desmielinização da substância branca, preservação das fibras subcorticais em U |
 |
 |
 |
 |
||
| Lobo occipital, Loyez. Idem | Lobo occipital, Sudan black. Perda da mielina da substância branca, preservação das fibras subcorticais. Acúmulos de macrófagos com material sudanófilo (setas) | Sudan black. Macrófagos com material sudanófilo fagocitado e axônios em degeneração na substância branca occipital |
 |
 |
 |
| Lobo occipital, Oil red O. Idem, macrófagos com lípides neutros no citoplasma | Ponte,LFB-Nissl. Desmielinização da via piramidal na base da ponte (áreas claras) | Bulbo,LFB-Nissl. Desmielinização completa das pirâmides (via córtico-espinal) |
 |
 |
 |
| Leucodistrofia
de Alexander.
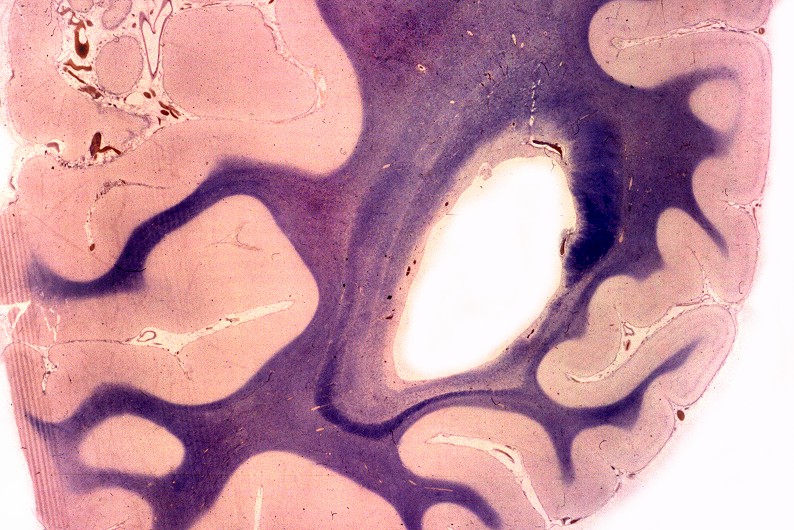
A doença de Alexander, descrita em 1949, é uma doença autossômica dominante rara, mas mutações novas são tão comuns que o padrão de herança se aproxima do de uma doença esporádica. Há formas infantil, juvenil e adulta. Na forma infantil o início é no 1º. ou 2º. ano de vida com óbito em 1 a 10 anos. Crianças mostram retardo no desenvolvimento neuropsicomotor, megalencefalia, convulsões, espasticidade e quadriparesia. Não há testes laboratoriais que permitam o diagnóstico, que só é possível em exame histológico. A doença é devida a mutações no gene para proteína glial ácida fibrilar (GFAP), o filamento intermediário próprio dos astrócitos, localizado no cromossomo 17q21-31. Há produção excessiva de GFAP, levando à formação de abundantes fibras de Rosenthal, e desmielinização difusa ou focal secundária. Radiologicamente, há lesões desmielinizantes simétricas com predomínio frontal. Pode haver degeneração cística, alargamento dos giros e tumefação da substância branca. Fibras arqueadas em U subcorticais não são poupadas. Há relativa preservação do córtex cerebral, cerebelo e tronco cerebral. Não há envolvimento de nervos periféricos ou outros órgãos. Na microscopia chama a atenção o grande acúmulo de fibras de Rosenthal, especialmente em localizações subpial, subependimária e perivascular. Astrócitos reacionais podem mostrar grânulos hialinos no citoplasma, correspondendo a fibras de Rosenthal em miniatura. Há extensa perda das baínhas de mielina da substância branca cerebral, por vezes chegando a cavitação. Em microscopia eletrônica, as fibras de Rosenthal são formadas por material homogêneo amorfo e eletrodenso, circundado por filamentos intermediários com 10 nm de espessura. Para ver exemplos em astrocitoma pilocítico, clique. Em imunohistoquímica, a periferia das fibras de Rosenthal é positiva para alfa-B-cristalina, GFAP, ubiquitina e proteína de choque térmico 27 Kd. |
| Neuropatologia. Caso 1. | ||
| Macro. Macrocefalia, caráter amolecido, friável da substância branca, dilatação ventricular | Hemisférios cerebrais, Loyez. Palidez da mielina | Bulbo, Loyez. Finas granulações correspondem a fibras de Rosenthal |
 |
 |
 |
| Bulbo, HE. Grande abundância de fibras de Rosenthal | Substância branca temporal, Loyez. Abundantes fibras de Rosenthal e escassez de fibras mielínicas | Bulbo, PTAH. Fibras de Rosenthal concentram-se em regiões subpiais e perivasculares. |
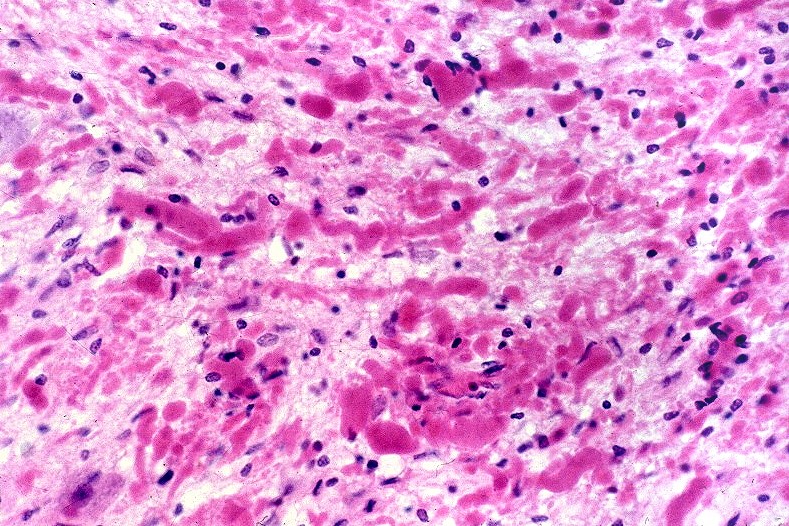 |
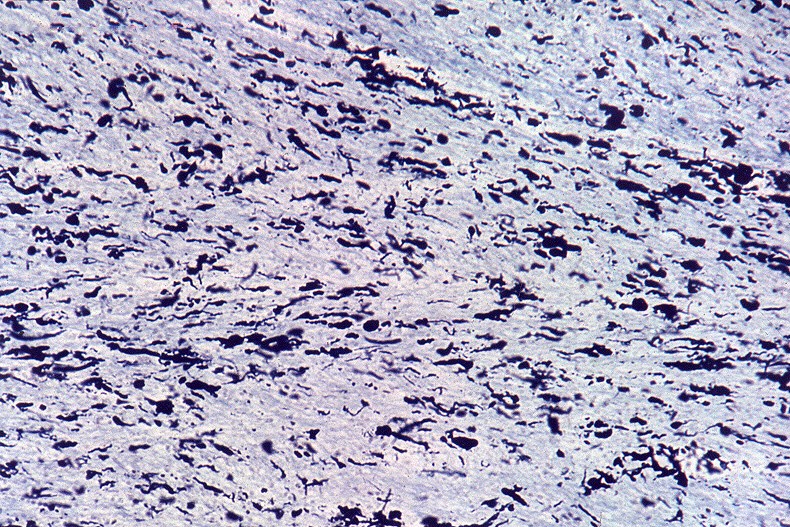 |
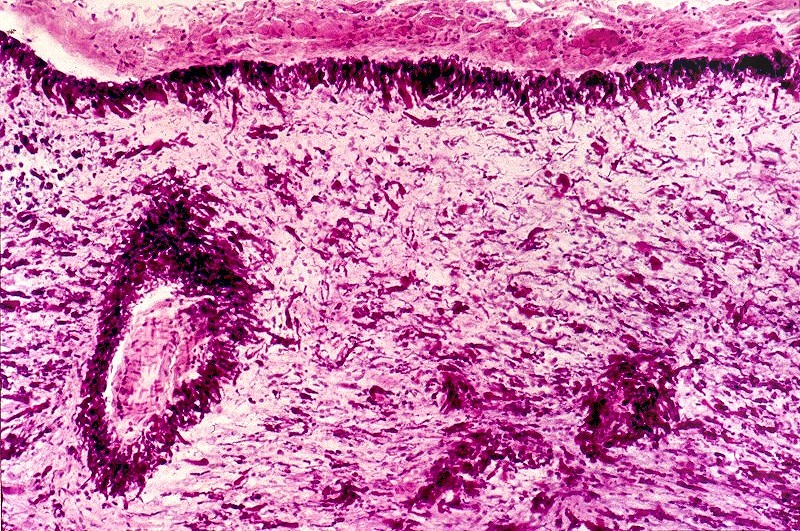 |
| Caso 2. | ||
| Ponte, LFB. Palidez dos fascículos piramidais na base da ponte | Bulbo, LFB. Palidez e atrofia das pirâmides (setas) | Medula cervical, LFB. Palidez dos fascículos piramidais nos funículos laterais |
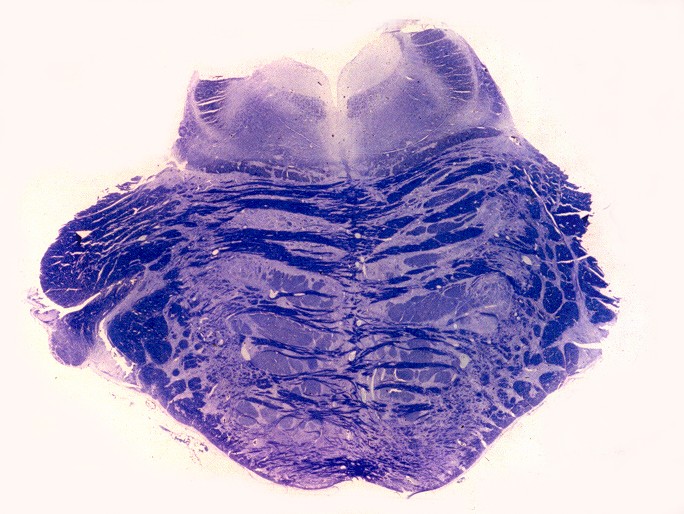 |
 |
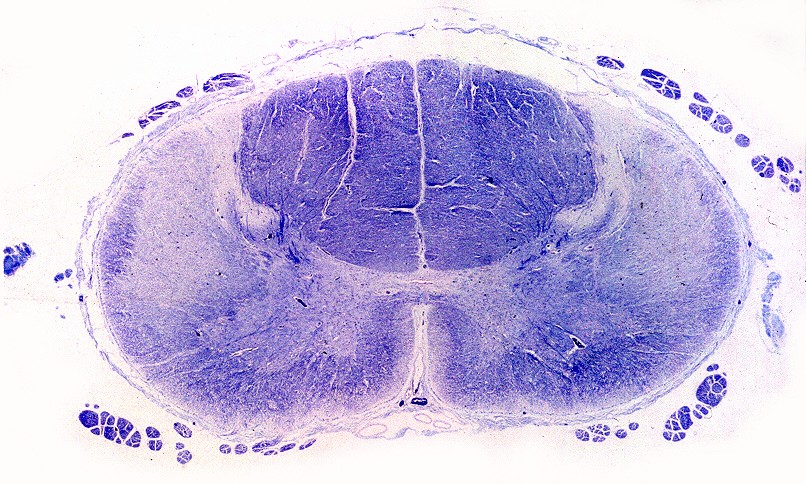 |
| Cerebelo, LFB. Palidez da mielina no hilo do núcleo denteado | Idem, detalhe das fibras mielínicas rarefeitas e com dilatações | |
 |
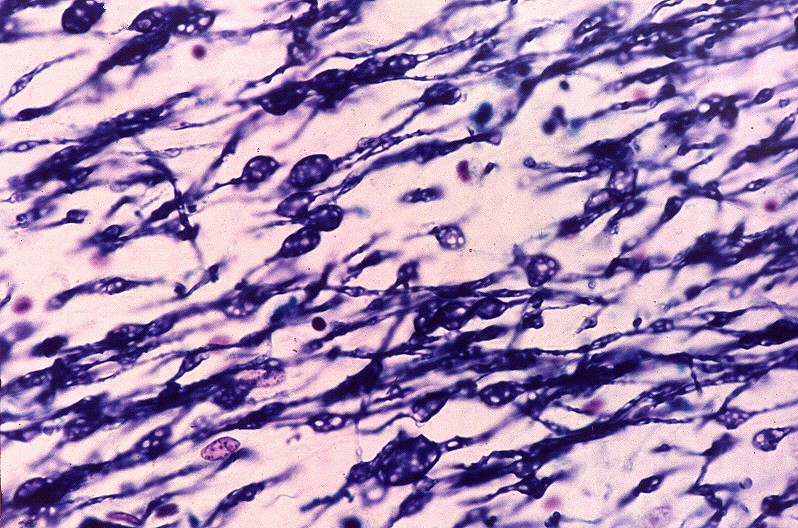 |
|
| Substância branca cerebral, Loyez. Fibras de Rosenthal (em negro) coexistindo com escassas fibras mielínicas remanescentes. | Idem, PTAH. | Medula cervical, LFB. Idem. |
 |
 |
 |
| Leucodistrofia
de Canavan.
Doença
autossômica recessiva, com gene localizado no cromossomo 17p13,2,
que codifica a enzima aspartato-acilase. Esta é necessária
para o catabolismo do N-acetilaspartato (NAA), e sua falta leva ao acúmulo
deste. Há predomínio em populações judias.
A freqüência de portadores de gene é 1 : 37,7 e a de
doentes 1:5000. O início dos sintomas é quase sempre
no período pós-natal, entre 10 semanas e 4 meses. Excepcionalmente
afeta crianças mais velhas. Há perda da fixação
e acompanhamento visual, dificuldade na sucção, irritabilidade,
perda do controle da cabeça (por atonia da musculatura cervical),
macrocefalia e convulsões. Sobrevida de meses a anos.
|
| Radiologicamente,
há desmielinização difusa e simétrica da substância
branca dos hemisférios cerebrais, com hipossinal em T1 e hipersinal
em T2, envolvimento das fibras em U e relativa preservação
da cápsula interna. Espectroscopia de prótons revela
aumento do NAA cerebral. Em um estudo, excreção urinária
de NAA foi mais de 200 vezes maior que em controles normais. NAA também
aumenta no plasma e líquor.
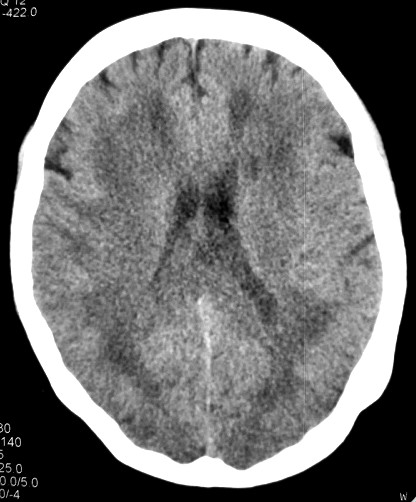
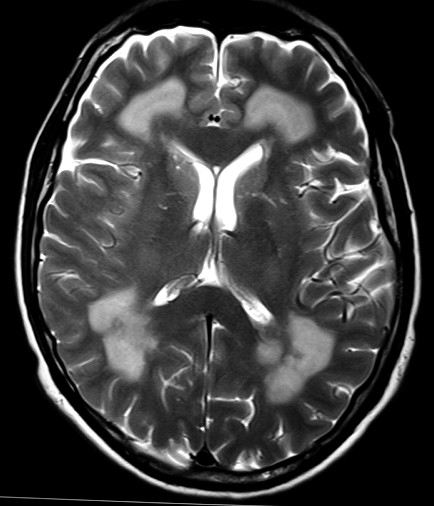
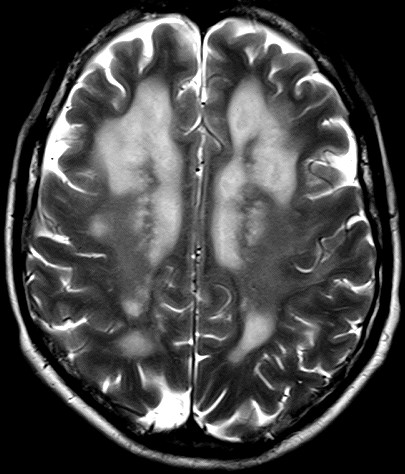
Masc. 43 a. Caso com biópsia (abaixo) mostrando as feições neuropatológicas habituais na doença de Canavan. |
|||
| TC sem contraste | T2 | ||
 |
 |
 |
 |
| T1 COM CONTRASTE | FLAIR | ||
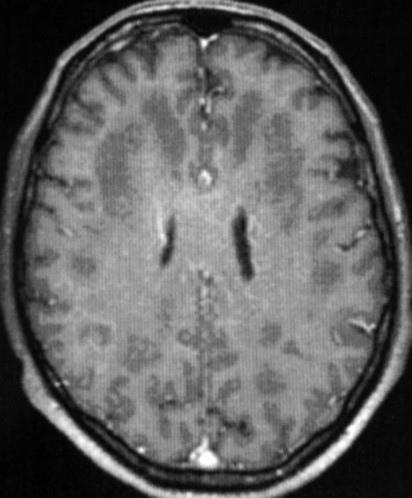 |
 |
 |
 |
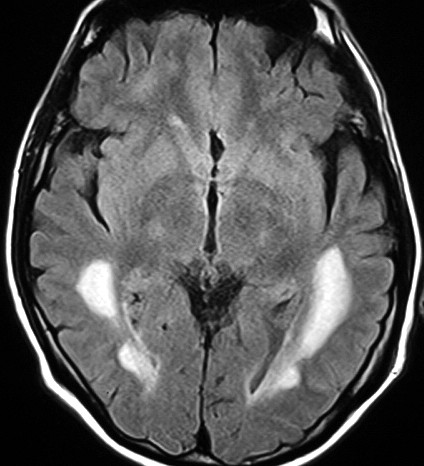
| Leucodistrofia de Canavan. Lâmina escaneada. HE. | 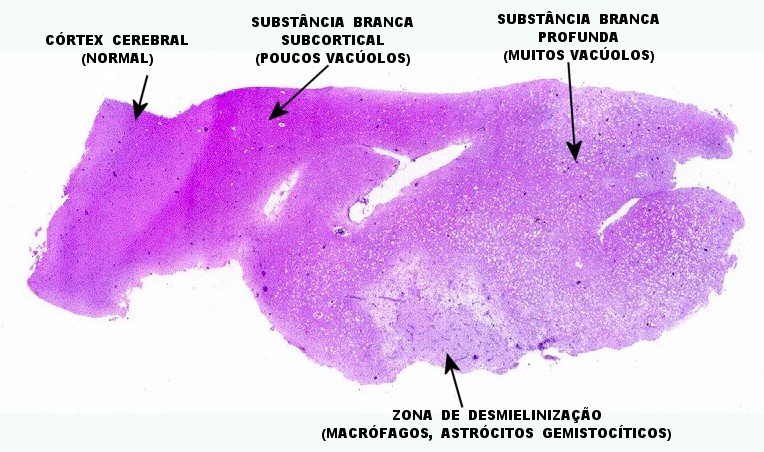 |
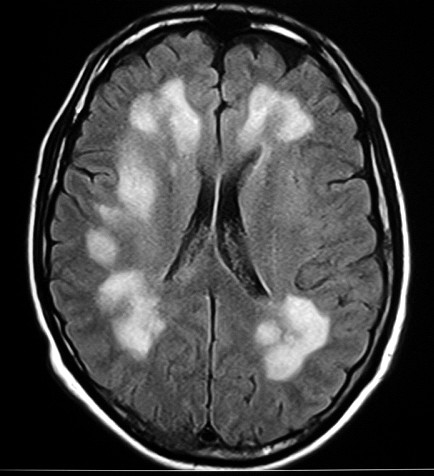
| Idem, LFB-Nissl. |  |
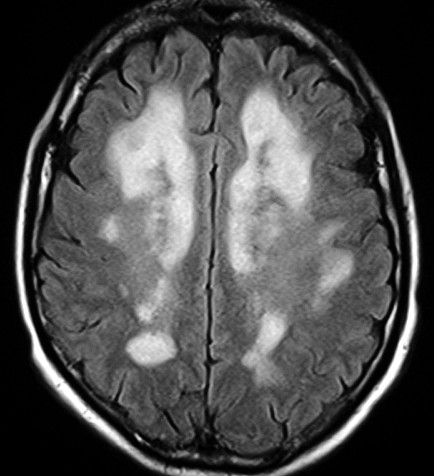
| Tentativa
de correlação
entre a lâmina escaneada e um corte axial pesado em T2. O córtex e a substância branca subcortical apresentam intensidades de sinal normais. A área profunda microvacuolada aparece com hipersinal, indicando maior hidratação. |
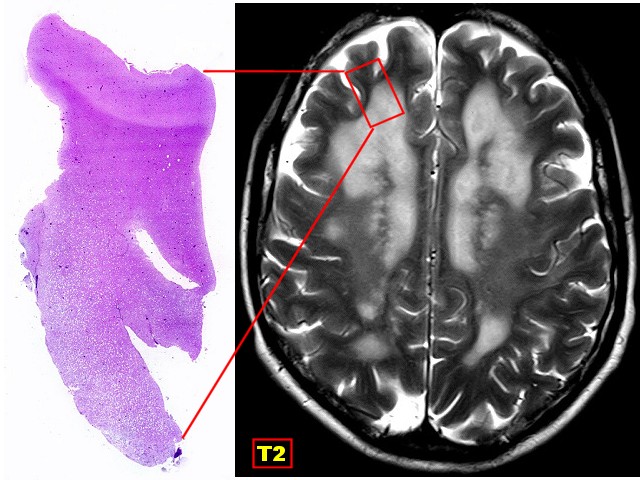 |
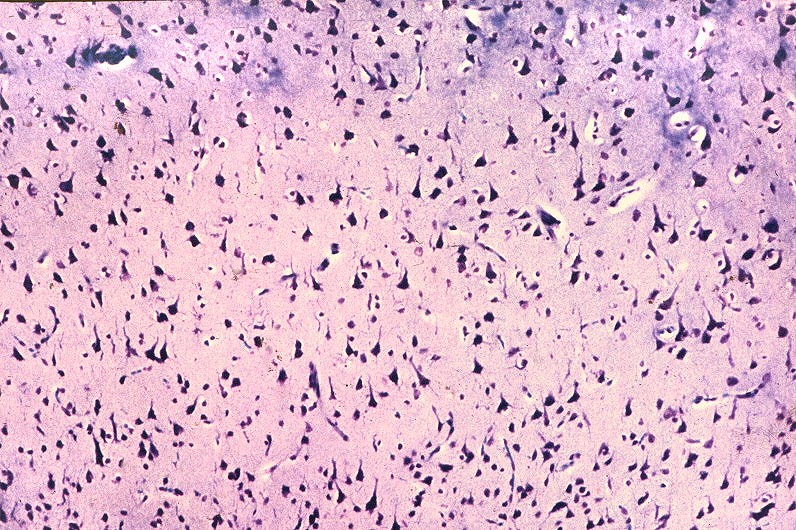
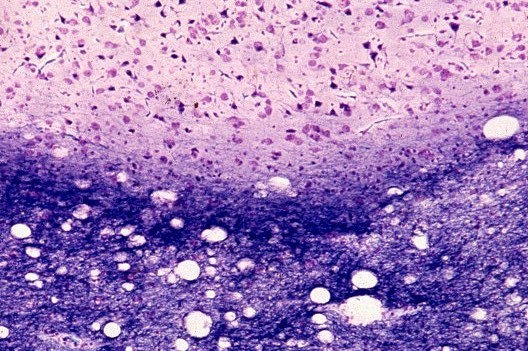
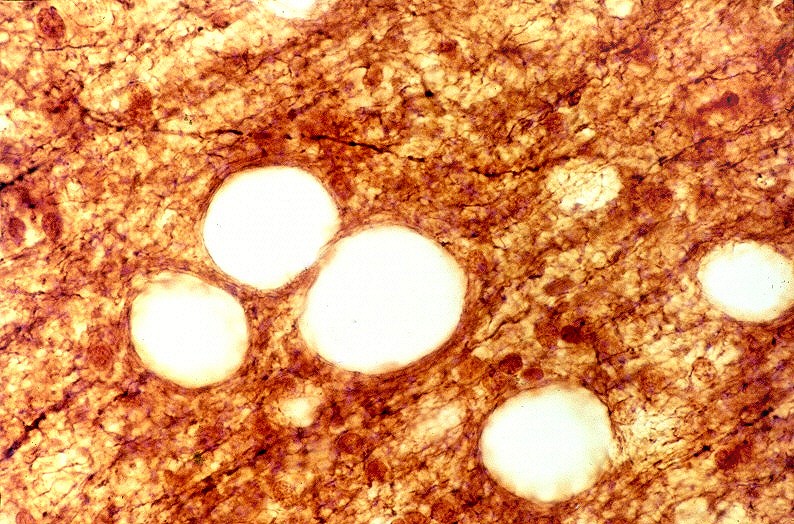
| Leucodistrofia de Canavan, Neuropatologia. Há megalencefalia nos estágios iniciais e microcefalia nos avançados. Na microscopia, a feição característica é a vacuolização disseminada na substância branca, especialmente na junção córtico-subcortical. A substância branca profunda mostra severa perda da mielina. Os axônios e oligodendrócitos são relativamente preservados. Há gliose astrocitária discreta e macrófagos são virtualmente ausentes. Em microscopia eletrônica, observa-se que os vacúolos são devidos à dissociação das lamelas da mielina ao nível da linha intraperiódica. | ||
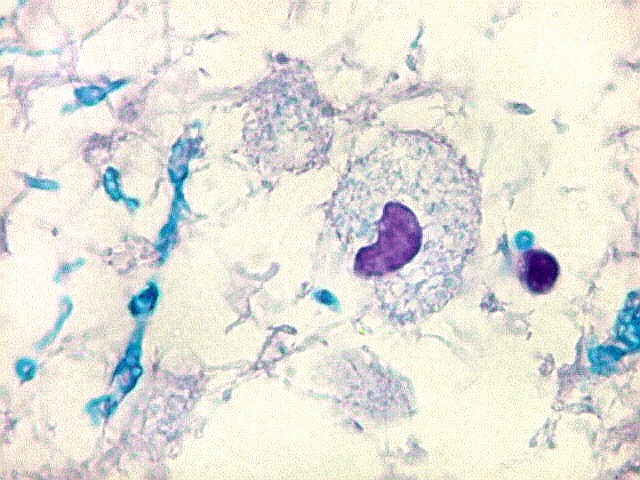
| HE. Vacúolos na substância branca. Transição entre área vacuolar e área desmielinizada. | Área desmielinizada. Macrófagos xantomatosos. | Área desmielinizada. Astrócitos gemistocíticos. |
 |
 |
 |
| LFB-Nissl. Lâminas escaneadas. Vacúolos na substância branca em meio a bainhas de mielina preservadas. | Área desmielinizada. Macrófagos xantomatosos. Rarefação ou desaparecimento da mielina | Área desmielinizada. Astrócitos gemistocíticos. |
 |
 |
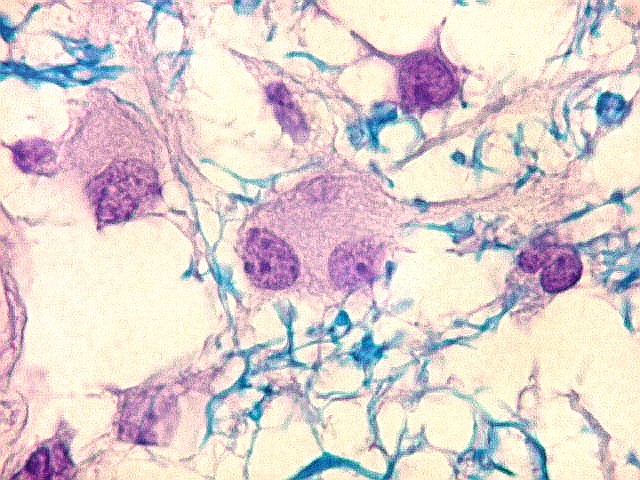 |
| VIM. Astrócitos gemistocíticos | VIM. Macrófagos xantomatosos | NF. Axônios na substância branca entre os vacúolos |
 |
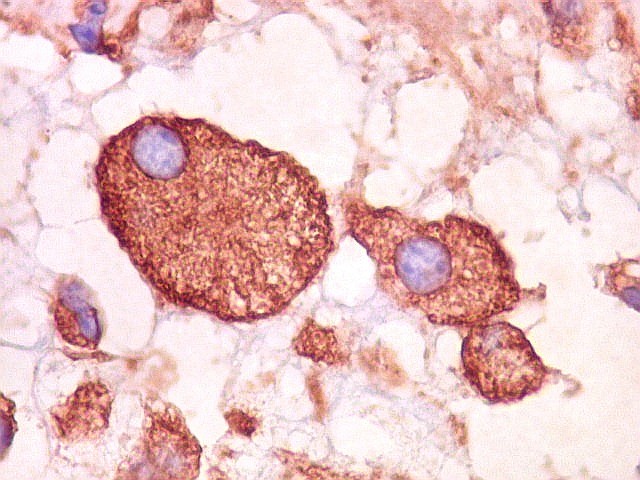 |
 |
| NF. Axônios persistem na área desmielinizada | NF. Neurônios normais no córtex cerebral | SNF. Glóbulos sinaptofisina positivos na área desmielinizada, presumivelmente em axônios. Axônios em contas de rosário |
 |
 |
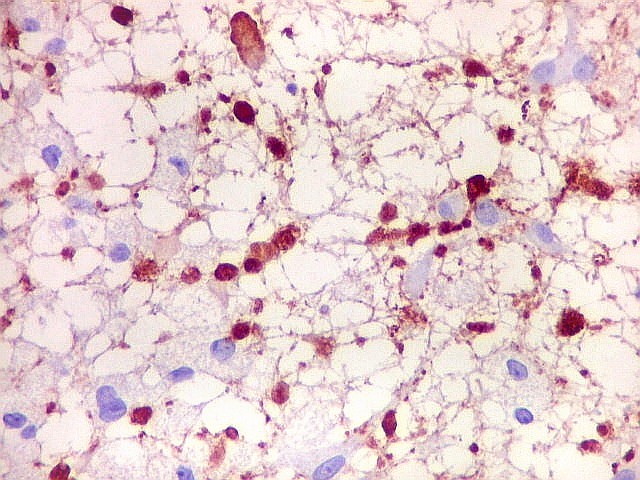 |
| Cromogranina. Idem. Axônios em contas de rosário | CD68. Macrófagos xantomatosos na área desmielinizada | CD68. Micróglia na substância branca com vacúolos |
 |
 |
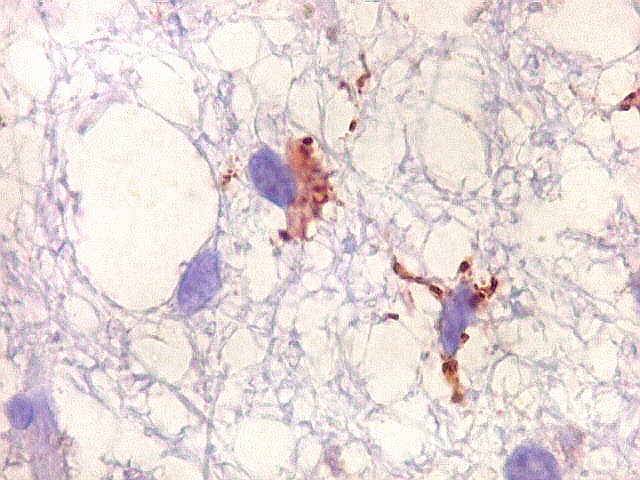 |
| Leucodistrofia de Canavan. Caso de arquivo. | ||
| HE. Córtex cerebral normal | Substância branca com vacúolos entre axônios e núcleos de células da glia | LFB-Nissl. Córtex cerebral normal |
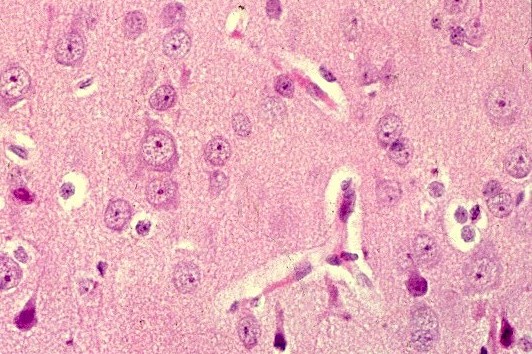 |
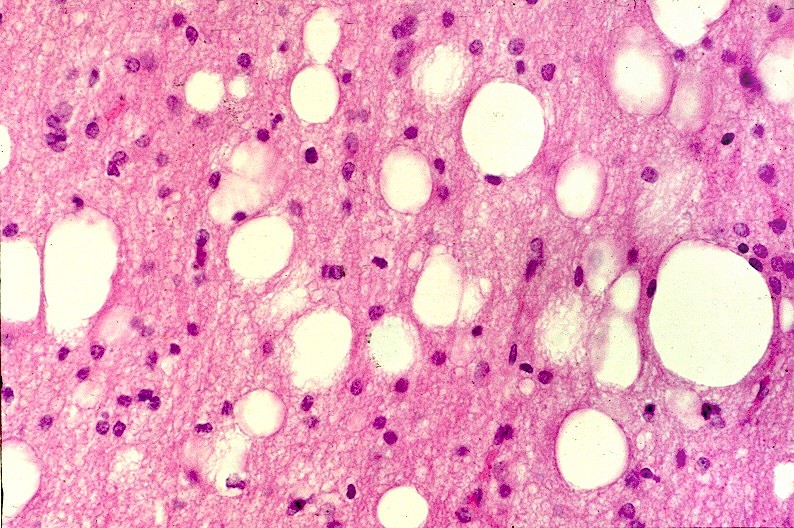 |
 |
| LFB-Nissl. Transição córtex - substância branca | Substância branca com vacúolos desviando axônios mielínicos | Glees. Vacúolos na substância branca, pobreza de axônios |
 |
 |
 |
| Leucodistrofia
de van der Knaap ou Leucoencefalopatia megalencefálica
com cistos subcorticais e alterações clínicas suaves).
Esta doença autossômica recessiva é caudada por mutações no gene MLC1 no cromossomo 22qtel, que codifica uma presumível proteína de membrana. A doença é notável pelo contraste entre a sintomatologia neurológica discreta e anormalidades radiológicas severas. Pode haver macrocefalia no primeiro ano de vida. Mais comumente, os sintomas apresentam-se o segundo ano de vida por retardo no desenvolvimento neuropsicomotor e lenta deterioração neurológica, disartria e ataxia. O perímetro cefálico estabiliza após o primeiro ano e depois segue a curva normal. A maioria dos pacientes necessita cadeira de rodas aos 8 a 10 anos, mas alguns conseguem andar curtas distâncias sem suporte até os 12-13 anos. Uma minoria desenvolve crises convulsivas controláveis por medicação. Deterioração cognitiva lentamente progressiva se instala a partir do meio da infância. Estudos patológicos mostram uma mielinopatia vacuolizante em que as camadas externas das baínhas de mielina estão delaminadas, mas as internas não são afetadas. Os axônios estão preservados. Estudos de imagem são notáveis pela ausência quase completa de mielina na substância branca subcortical. A substância branca central é poupada, particularmente no corpo caloso e lobos occipitais. A substância branca periférica parece tumefeita, com alargamento dos giros. Uma feição característica são cistos subcorticais nos lobos frontais e temporais. A substância branca cerebelar é menos afetada, mas pode haver hipersinal em T2 nos casos mais graves. Os aspectos
radiológicos são semelhantes aos da doença de Canavan,
sendo a diferenciação baseada em critérios clínicos
(início no primeiro ano na doença de Canavan, segundo ano
na doença de van der Knaap); envolvimento do globo pálido
e tálamo quase sempre presente na doença de Canavan, ausente
na doença de van der Knaap; espectroscopia de prótons mostrando
grande pico de NAA no Canavan, pequeno ou ausente na doença de van
der Knaap, e restrição à difusão reduzida
no Canavan, aumentada na van der Knaap.
|
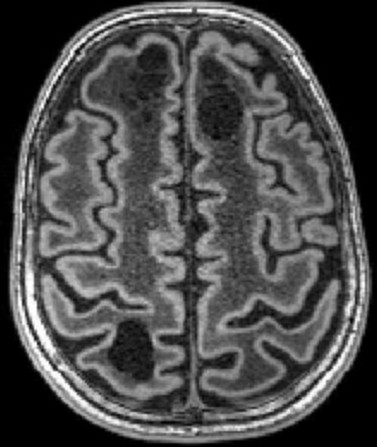
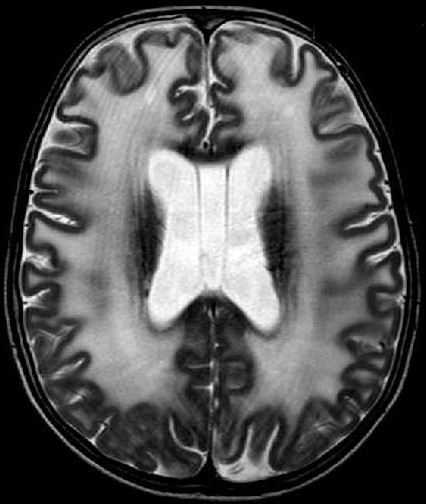
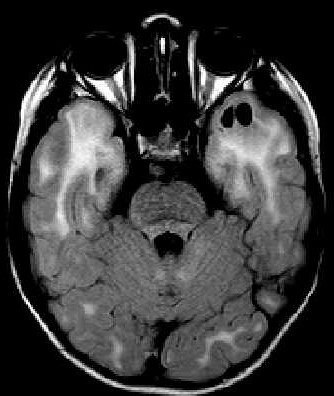
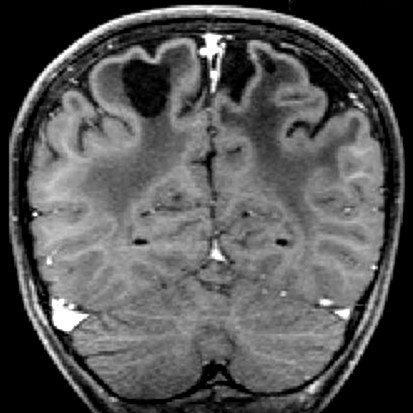
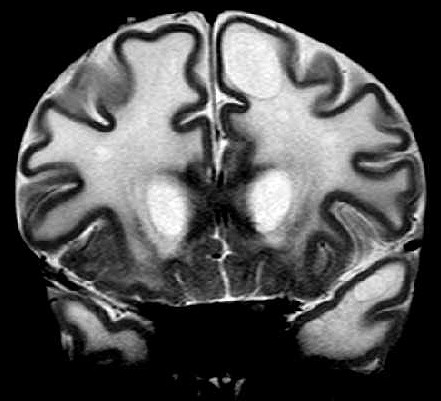
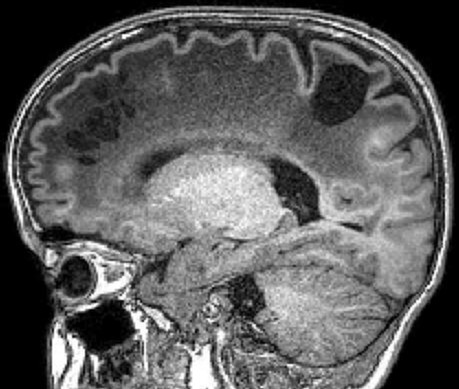
| Masc. 11 a 5 m. Paciente acompanhado desde idade de 8 meses. Clique para história clínica, destaques e detalhes de 4 RMs. Abaixo fotos do exame mais recente (2014) mostram intensa hipomielinização difusa da substância branca dos hemisférios cerebrais, com cistos subcorticais múltiplos, cujo tamanho vem aumentando ao longo da evolução. Também dilatação dos ventrículos laterais, cavum septi pellucidi e cavum vergae. Córtex cerebral, estruturas cinzentas profundas, cerebelo e tronco preservados. | |||
| T1 SEM CONTRASTE | T2 | FLAIR | |
 |
 |
 |
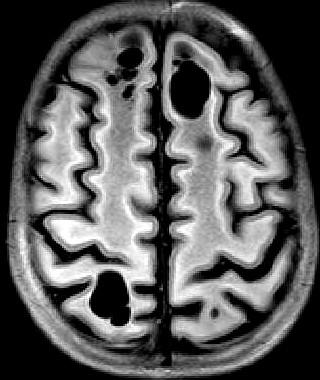 |
| T1 SEM CONTRASTE | T2 | T1 SEM CONTRASTE | |
 |
 |
 |
 |
| Leucodistrofia
associada a distrofia muscular congênita por deficiência de
merosina.
Distrofias musculares congênitas são um grupo heterogêneo caracterizadas por hipotonia, fraqueza, freqüentemente contraturas congênitas (artrogripose) e alterações distróficas do músculo em biópsias. Parte dos pacientes tem anormalidades confinadas aos músculos, mas em outros também há anormalidades nos olhos e cérebro, podendo ser divididos em 3 grupos:
A ressonância magnética mostra retardo da mielinização ou hipomielinização na substância branca central do cérebro. Pode haver discreta hipoplasia do vermis cerebelar e da ponte. O diagnóstico é feito pela combinação destes achados com a biópsia muscular, associados aos dados clínicos. O motivo das alterações da mielina não é claro. Há evidência recente de que a falta de laminina altera as vias bioquímicas que promovem a maturação dos oligodendrócitos. Relucio et al., 2009. Laminina. Glicoproteína de múltiplos domínios, formada por 3 cadeias polipeptídicas (alfa, beta e gama) associadas por pontes dissulfídicas na forma de uma cruz assimétrica. A haste vertical da cruz é formada pela cadeia alfa. As cadeias beta e gama enrolam-se em espiral em torno da parte inferior da haste e formam cada uma um dos braços da cruz. A laminina é componente essencial da membrana basal em associação com outras moléculas, como colágeno IV, perlecan e nidogen. Os receptores na membrana celular para as moléculas da membrana basal são da família das integrinas. Outro importante receptor (ponto de ancoragem) para a laminina é a proteína transmembrana distroglican que, juntamente com as integrinas, ajuda a organizar a montagem da membrana basal. O termo merosina é usado para a laminina 2. S-merosina é sinônimo de laminina 4. A cadeia alfa-2 entra na constituição das lamininas 2 e 4. |
| Masc. 5 a. Paciente com distrofia muscular congênita (biópsia a seguir) e alterações na substância branca compatíveis com leucodistrofia. Clique para história clínica e exames de imagem em detalhe. | |||
| TC sem contraste | T2 | ||
 |
 |
 |
 |
| T1 SEM CONTRASTE | FLAIR | ||
 |
 |
 |
 |
| Biópsia de músculo bíceps braquial com a idade de 6 anos, mostrando alterações distróficas. | |
 |
|
 |
 |
Fontes.
|
| Para
mais sobre leucodistrofias : Texto
ilustrado linkado
Para outras doenças desmielinizantes e degenerativas, ver também Neuroimagem, Neuropatologia, Banco de Imagens. |
|||
 |
 |
 |
 |
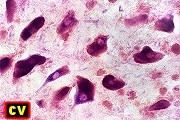
| Adrenoleucodistrofia. Foto - desmielinização da substância branca frontal com preservação das fibras em U | Leucodistrofia metacromática. Foto - armazenamento de substância metacromática (sulfátides) em neurônios do núcleo do facial na ponte | Leucodistrofia metacromática. Foto - armazenamento de substância metacromática em macrófagos na substância branca cerebelar | Leucodistrofia sudanófila. Foto - desmielinização e cavitação da substância branca frontal, e dilatação ventricular |
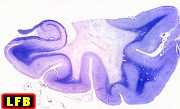 |
 |
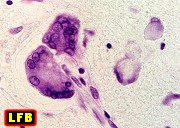 |
 |
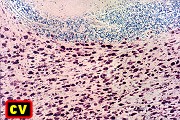
| Leucodistrofia sudanófila. Foto - desmielinização da substância branca temporal com preservação das fibras em U | Leucodistrofia de células globóides - 3 casos. Foto - desmielinização da substância branca occipital com preservação das fibras em U | Leucodistrofia de células globóides. Foto - células globóides (macrófagos) na substância branca cerebelar | Leucodistrofia de Pelizaeus - Merzbacher. Foto - Aspecto tigróide da substância branca |
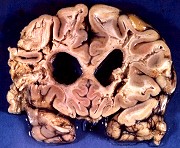 |
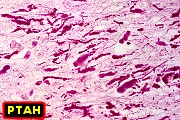 |
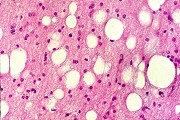 |
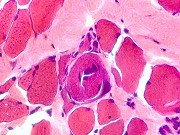 |
| Leucodistrofia de Alexander. Foto - megalencefalia e aspecto friável da substância branca | Leucodistrofia de Alexander. Foto - abundantes fibras de Rosenthal na medula lombar | Leucodistrofia de Canavan. Foto - vacúolos na substância branca | M. 5 a. Distrofia muscular congênita por deficiência de merosina, associada a leucodistrofia. Foto - biópsia muscular |
| LEUCODISTROFIAS .- Casos de neuroimagem | |||
 |
 |
 |
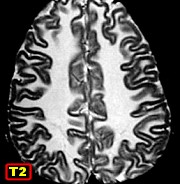 |
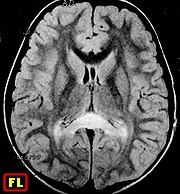
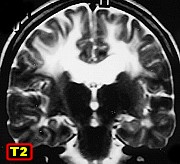
| M. 6 a. Adrenoleucodistrofia | M. 11 a. Adrenoleucodistrofia | M. 10 a. Adrenoleucodistrofia | M. 7 a. Leucodistrofia metacromática |
 |
 |
 |
 |
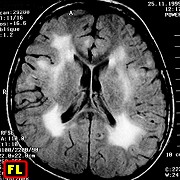
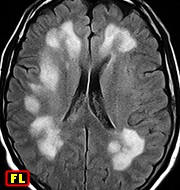
| F. 30 a. Provável leucodistrofia metacromática, forma do adulto | F. 56 a. Provável leucodistrofia metacromática, forma do adulto | M. 5 a. Leucodistrofia associada a distrofia muscular congênita por deficiência de merosina | M. 11 a. Doença de van der Knaap (leucoencefalopatia megalencefálica com cistos subcorticais). Acompanhamento de 11 anos. Melhores imagens de 4 exames |
| Exames em detalhe | |||
| Caso com correlação neuropatológica | |||
 |
 |
 |
|
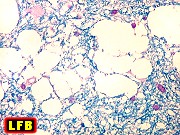
| M. 43 a. Leucodistrofia com alteração vacuolar na substância branca lembrando a doença de Canavan | HE, LFB-Nissl | Imunohistoquímica | |
| Módulo Neuro - Página Inicial | Outros módulos | Técnicas histológicas | e-mails : [email protected]_
__[email protected] |
|
|
|
|